2 月 11 日,世界卫生组织(WHO)正式将新冠肺炎病命名为 COVID-19,国际病毒分类委员会(ICTV)将新冠病毒命名为 SARS-CoV-2。SARS-CoV 病毒引起 SARS 病,SARS-CoV- 2 病毒引起 COVID-19 病,从官名上能看出新冠病毒与非典病毒有着密切联系,科研人员也揭示了不同病毒间蛋白质(氨基酸序列)的差异可能是新冠病毒比 SARS 和 MERS 更具传染性的原因。除了蛋白方面的差异,病毒基因组也必然存在着差异。从 2003 年的 SARS 到 2012 的 MERS,再到 2020 的 COVID-19,毫无疑问,我们肉眼看不到的冠状病毒也在发生着变异与进化,这些变异有利于病毒自身,却危害人类生命健康。今天小编要分享的便由变异所起——拷贝数变异的相关信息,提供拷贝数变异检测方面的参考。
拷贝数变异的形成和致病机制
拷贝数变异属于结构变异的一种,可分为拷贝数增加和拷贝数减少。拷贝数变异是人类遗传多样性的来源之一。而且通过重组、复制或其他方式产生的拷贝数变异似乎比单核苷酸多态性要高得多。但拷贝数变异并不一定会导致疾病,可能仅仅是作为一种多态性而存在,这与大量的单核苷酸多态性一样,是良性的。异常的拷贝数变异通常是癌症、遗传病甚至是某些复杂疾病的重要分子机制。
拷贝数变异的产生主要涉及 4 种机制:
非同源重组、非同源连接、L1 反转录以及复制叉滞后连接等。
拷贝数变异对表型的影响机制主要有:
1)基因剂量;
2)功能失活;
3)基因融合;
4)位置效应;
5)横向效应等。
拷贝数变异与三体综合征
人类很早就认识到拷贝数变异与疾病之间的关系,但受到检测精度的限制,早期的发现以大的染色体水平的变异为主。随着基因芯片、高通量测序等多种检测技术的快速发展,越来越多的拷贝数变异被检测出来,包括亚显微结构的拷贝数变异检测成为可能。
三体综合征是典型的基因组拷贝数变异引起的疾病,例如 21 三体综合征、18 三体综合征和 13 三体综合征。21 三体综合征又名唐氏综合征,发病率约为 1 /700。13 三体综合征于 1657 年被 Thomas Bartholin 初次记载,并由 Klaus Patau 在 1960 年报道染色体核型,也称为 Patau 综合征。18 三体综合征于 1960 年被 John Hilton Edwards 报道,也被称为 Edwards 综合征。
拷贝数变异数据库
拷贝数变异数据收录于 DGV 数据库(http://dgv.tcag.ca/dgv/app/home),记录了一系列基因变异与表型相关的信息,数据库信息持续更新,截至 2020.2.19,DGV 数据库收录了总共 72 项研究 6359956 个样本的 CNV 信息,大多数的 CNV 分布在 1 -10Kb,其次为 100-500bp。
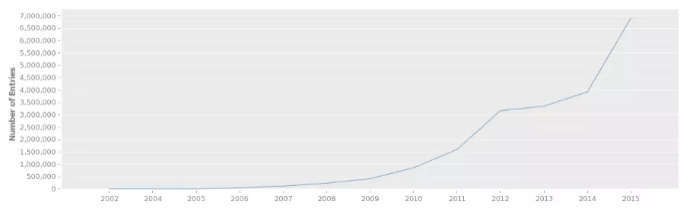
图 1 DGV 数据库变异数目逐年增加情况
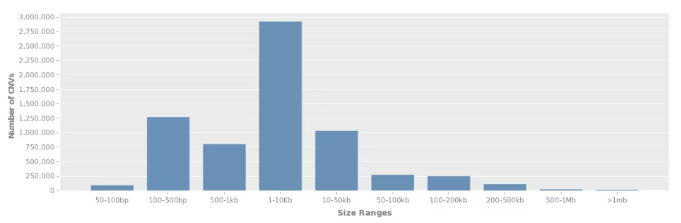
图 2 DGV 数据库 CNV 大小的分布情况
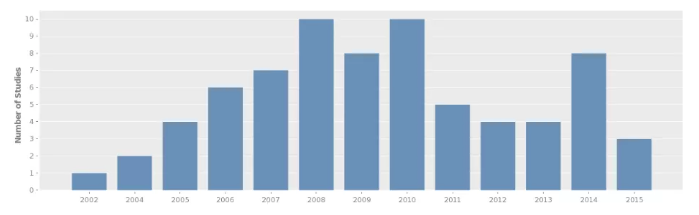
图 3 DGV 数据库项目数情况
拷贝数变异检测平台的技术比较
传统的染色体核型分析、全基因组范围内 CNV 芯片、高通量测序技术均可以对 CNV 进行检测,接下来的两篇文献均是关于芯片和测序两大 CNV 检测平台的比较研究,孰优孰劣让我们一探究竟。
文献 1 是 2016 年香港中文大学医学院团队发表于国际医学遗传学权威学术杂志 Genetics in Medicine 的文章讨论了 NGS 技术是否可以作为常规临床应用中 CNV 检测的替代方法。

该团队使用低覆盖度的全基因组测序流程,对 570 名患者的多中心组进行了全基因组 CNV 分析(> 50 kb)和芯片检测分析。根据美国医学遗传学和基因组学指南对 CNV(即致病性 CNV,pCNV)进行了分类。结果发现,CMA 和基于 NGS 的方法在 71 个验证样本中的 pCNV 的结果一致(如下图表)。
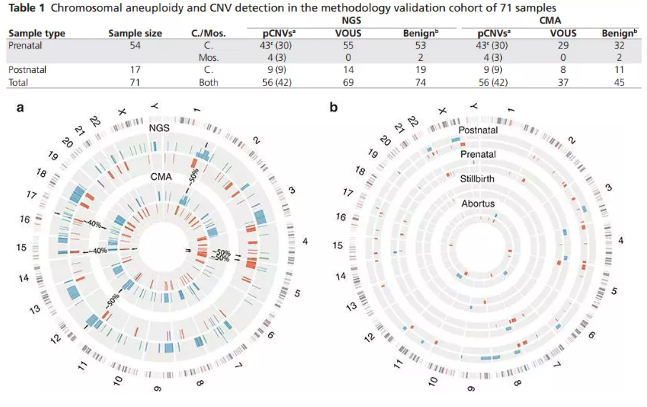
研究者也指出,高深度全基因组测序(whole genome sequencing,WGS)可以提供整个基因组的全面视图,是一种高通量的检测单核苷酸变异、插入 / 缺失、拷贝数变异和基因组结构重组的技术手段。但目前而言,高深度 WGS 的价格高昂,且变异信息较多,较难在产前诊断中广泛应用;而基于低深度 WGS 的水平,能够在降低每个样本的测序成本基础上,同时提高检测通量,达到低深度高通量的目的(low-pass WGS)。
文献 2 也是来自于同一团队,2019 年 12 月份发表于 Genetics in Medicine,增加了样本例数,对低深度高通量全基因组测序和染色体微阵列分析进行了深入比较,分析结果支持了将低深度全基因组测序用于产前诊断中以提供更多有意义的临床信息。

研究人员将 2016-2019 年入组的 1023 名孕妇同时进行了 CMA 和 NGS 检测。CMA 检测出共 121 例样本的异常,包括 87 个非整倍性和 37 个致病或可能致病的 CNV,这两种染色体异常存在部分重叠;而低深度测序不仅检测到了全部的 CMA 检出的染色体异常,还额外检测到 17 例非整倍性或致病 / 可能致病的染色体拷贝数变异。作者进一步分析了测序分析结果的灵敏度和特异性,以芯片分析为基准,低深度全基因组测序达到了 99.9%(121/121)的灵敏度和 87.7%(121/138)的特异性,总体诊断阳性率为 13.5%(138/1023)。在 DNA 起始量方面,芯片起始量 300ng DNA 和低深度测序起始量 50ng 为标准进行了样本制备,结果是低深度全基因组测序样本中有 5 例未能通过初期试验,而芯片则有 47 例未通过,也就是造成了重复实验率分别为 0.5%(5/1023)和 4.6%(47/1023),这些结果综合说明低深度全基因组测序应用于产前诊断的样本要求更低,实验也更加稳定。
从 CNV 检测技术总体来看,传统的染色体核型分析一直被认为是确诊染色体变异的标准,也是染色体病产前诊断的一线方法,但是检测周期长、分辨率较低,无法检出 5Mb 以下的 CNV。目前主要用于全基因组范围内 CNV 检测的“金标准”技术为 CNV 芯片,检测范围广、分辨率较高;缺点是成本较高,在大规模的产前诊断应用中有所限制。而近年来发展起来的高通量测序技术,为 CNV 检测提供了新的手段,具有成本低、通量高、检测便捷等优势。
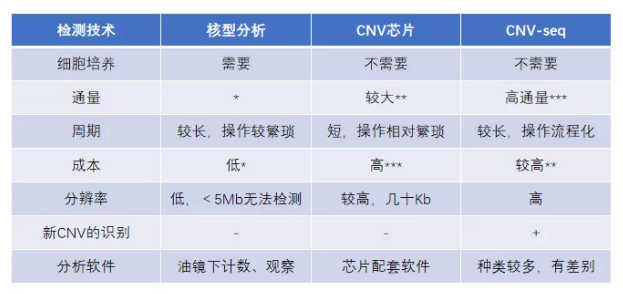
不可否认,芯片方法依然是拷贝数检测的金标准,基于高通量测序平台检测 CNV 的多篇文献中也提到了这一点,并以此为基准分析测序方法的准确度和灵敏度。高通量的方法作为后起之秀,检测结果依赖于测序质量、测序数据量、分析软件等,完全替代芯片还需些时间。伯豪生物提供芯片和高通量测量测序两种平台的检测服务,小编建议各位老师们可以按需选择。
这次疫情已导致 7 万余人受到新冠病毒的感染,同时得到了国内外千千万万人的高度关注,我们有理由相信,人类终将赢得这场战“疫”,而胜利之后的人类该会敬畏自然,善待自然界的其他动物们!

