信息时代就是好,没有自己的数据咱可以从免费的数据库找数据呀!目前 TCGA 和 GEO 数据库简直就是专为科研人员准备的免费的宝库。前面一直讲的空间转录组,今天来穿插着讲一节 TCGA & GEO 数据分析换换口味吧。
今天主要给大家介绍一下对转录组 TCGA & GEO 的数据进行分析,包括差异分析、富集分析、单基因展示以及其它个性化做图。除了想利用公共数据库挖掘转录组数据的发文章的同学之外,这篇教程其实特别适合自己做了少量转录组样本又不想做后期验证,想直接用公共数据库的数据来验证自己数据结果的朋友。
目前各种 TCGA & GEO 数据下载的教程太多了,所以今天直接省略这部分部分,直接从后面的分析开始。废话不多说,直接上干货!
一、准备文件
基因表达矩阵文件,可以是 TCGA 下载的转录组数据,也可以是 GEO 下载的转录组数据,格式如下(行是基因,列是样本):
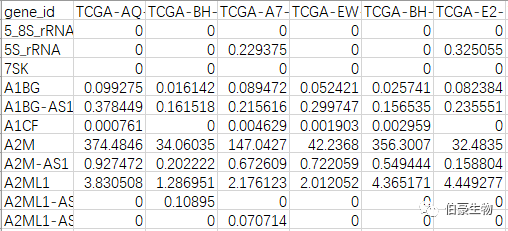
样本分组文件,格式如下(type 是分组):
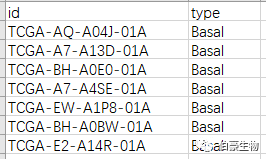
二、差异分析
一般 TCGA 或 GEO 上大量样本数据的差异分析 limma 用的比较多,所以这里也使用 limma 包来分析差异基因,注意需要先安装 limma R 包。
安装 limma 包:
if (!requireNamespace("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install("limma")
分析差异:
## 读取基因表达件,取 log2
geneexp = read.table("gene_exp_BRCA.txt",header=T,row.names=1,sep="\t")
geneexp = log2(geneexp+1)
## 读取分组文件
group_file = read.table("sample_group_BRCA.txt",header=T,row.names=1,sep="\t",as.is =TRUE)
rownames(group_file) = gsub('-','.',rownames(group_file))
geneexp = geneexp[,rownames(group_file)]
### 差异分组设置
samps<-factor(group_file$type)
design <- model.matrix(~0+samps) ;
colnames(design) <- levels(samps)
### 模型拟合
fit <- lmFit(geneexp, design)
cont.matrix<-makeContrasts(Basal-Normal,levels=design)
fit2 <- contrasts.fit(fit, cont.matrix)
fit2 <- eBayes(fit2)
final<-topTable(fit2, coef=1, number=dim(geneexp)[1], adjust.method="BH")
> head(final)
logFC AveExpr t P.Value adj.P.Val B
SDPR -4.358861 2.679133 -46.10828 8.888505e-122 4.738017e-117 267.4862
TPX2 4.030685 3.807811 45.39822 2.571983e-120 6.854979e-116 264.1584
UBE2C 4.089727 3.270516 43.72542 8.428935e-117 1.497681e-112 256.1483
CDC20 3.971916 3.450304 42.85468 6.270780e-115 8.356598e-111 251.8811
FIGF -3.220704 1.583802 -40.93757 1.053838e-110 1.123497e-106 242.2399
FAXDC2 -2.179867 1.637909 -39.05494 2.084335e-106 1.851758e-102 232.4283
三、绘制差异基因散点图和火山图
这一步需要用到 ggplot2 绘图,没有安装的话需要先安装 ggplot2 包。
# 绘制差异基因散点图和火山图
library(ggplot2)
g1 = "Normal"
g2 = "Basal"
g1_exp = geneexp[rownames(final),rownames(group_file)[which(group_file$type==g1)]]
g2_exp = geneexp[rownames(final),rownames(group_file)[which(group_file$type==g2)]]
g1_mean = apply(g1_exp,1,mean)
g2_mean = apply(g2_exp,1,mean)
type=rep('No',length(g1_mean))
type[which(final$logFC> 1 & final$adj.P.Val <0.05)] = "Up"
type[which(final$logFC < -1 & final$adj.P.Val < 0.05)] = "Down"
datam = data.frame(g1_mean,g2_mean,logFC=final$logFC,FDR=final$adj.P.Val,type,stringsAsFactors=FALSE)
## 散点图
ggplot(datam,aes(g1_mean,g2_mean,colour=type))+
geom_point(stat="identity",size=1)+theme(legend.title=element_blank())+scale_color_manual(values =c("Down"='blue',"No"='grey',"Up"='orange'))+
labs(x=paste(g1,'Log2(FPKM+1)'),y=paste(g2,'Log2(FPKM+1)'),title=paste(g2,'VS',g1,sep=""))+
coord_cartesian(ylim=c(0,10),xlim=c(0,10))+geom_segment(aes(x = 0, y = 0, xend = 10, yend = 10),size=1,colour="#999999",linetype="dotted")+theme(plot.title = element_text(hjust = 0.5),title=element_text(face="bold",size=15,colour="black"),axis.title=element_text(face="bold",size=13,colour="black"),axis.text.x=element_text(face="bold",size=12,colour="black"),axis.text.y=element_text(face="bold",size=12,colour="black"),legend.text=element_text(face="bold",size=13,colour="black"))
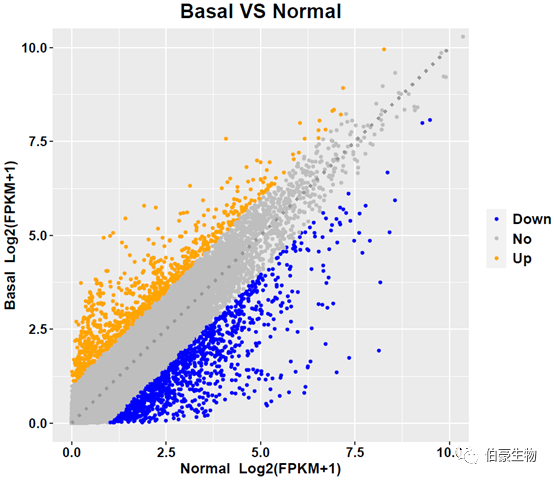
散点图结果如下:
## 绘制火山图
ggplot(datam,aes(logFC,-log10(FDR),colour=type))+
geom_point(stat="identity",size=1.2)+theme(legend.title=element_blank())+scale_color_manual(values =c("Down"='blue',"No"='grey',"Up"='orange'))+
labs(x="Log2 (FC)",y="-Lg10 (FDR)",title=paste(g2,'VS',g1,sep=""))+coord_cartesian(xlim=c(-5,5))+
geom_hline(aes(yintercept=1.3),colour="white",size=1.1)+
geom_vline(aes(xintercept =-1),colour="white",size=1.1)+geom_vline(aes(xintercept =1),colour="white",size=1.1)+
theme(axis.title.y = element_text(vjust=-0.1),axis.title.x = element_text(vjust=-0.6),title = element_text(vjust=0.8))+theme(plot.title = element_text(hjust = 0.5),title=element_text(face="bold",size=15,colour="black"),axis.title=element_text(face="bold",size=13,colour="black"),axis.text.x=element_text(face="bold",size=10,colour="black"),axis.text.y=element_text(face="bold",size=10,colour="black"),legend.text=element_text(face="bold",size=12,colour="black"))
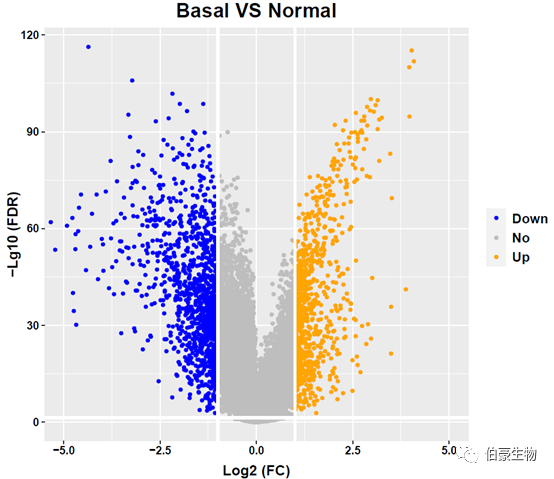
火山图结果如下:
四、差异基因富集分析
这里我们介绍一下怎么用 clusterProfiler R 包来做差异基因富集分析。需要先安装好 clusterProfiler,org.Hs.eg.db 两个 R 包。
if (!requireNamespace("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install("clusterProfiler")
BiocManager::install("org.Hs.eg.db")
GO 富集分析
library(clusterProfiler)
library(org.Hs.eg.db)
### 提取差异基因 list
diffgenes <- final[(final[,"adj.P.Val"]<0.05 & abs(final[,"logFC"])>=1),]
genelist <- diffgenes[,"logFC"]
names(genelist) = rownames(diffgenes)
##id 转换,将 SYMBOL 转换成 ENTREZID
gene = bitr(rownames(diffgenes), fromType = "SYMBOL", toType = "ENTREZID", OrgDb = "org.Hs.eg.db")
#####################GO 富集
ego <- enrichGO(
gene = gene$ENTREZID,
keyType = "ENTREZID",
OrgDb = "org.Hs.eg.db",
ont = "ALL",
pAdjustMethod = "BH",
pvalueCutoff = 0.05,
qvalueCutoff = 0.05,
readable = TRUE)
# 再用 setReadable 函数将基因 ID 映射到基因 Symbol
ego2 <- setReadable(ego, OrgDb = "org.Hs.eg.db", 'ENTREZID')
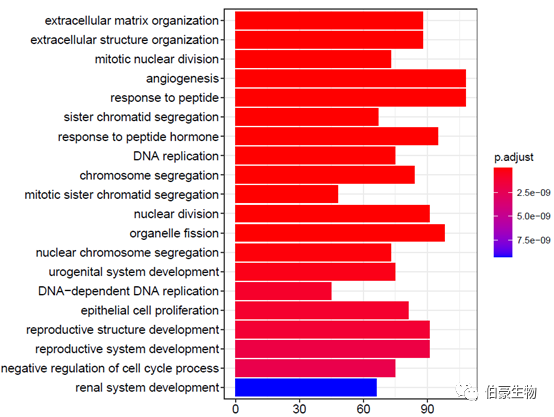
绘制柱状图:
barplot(ego2,showCategory = 20)
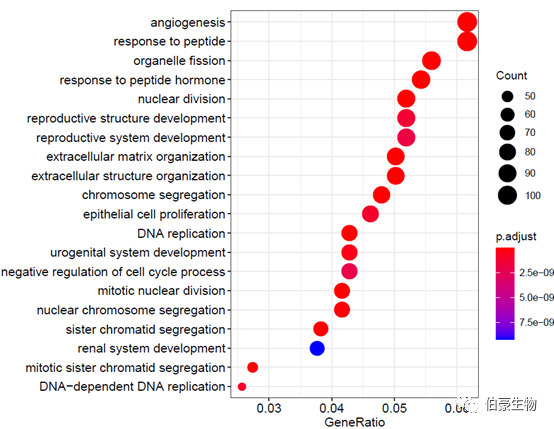
绘制气泡图:
dotplot(ego2, showCategory = 20)
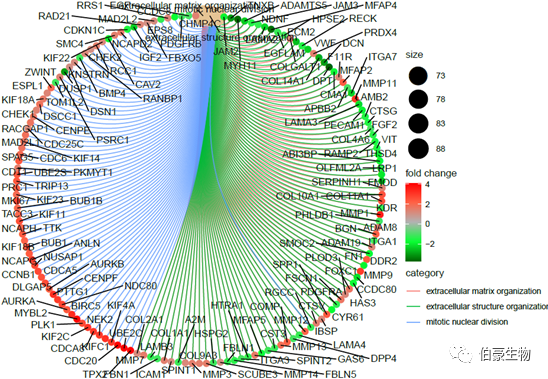
绘制网络图 1:
cnetplot(ego2, showCategory = 3,foldChange=genelist, circular = TRUE, colorEdge = TRUE)
绘制网络图 2:
emapplot(ego2, foldChange=genelist, showCategory = 20)

KEGG 富集分析
注意这一步需要连接网络,因为 clusterProfiler 是在线抓取新的 pathway 数据库的。当然也有用 kegg.db R 包直接内置数据库的情况,这里不做介绍。
######################KEGG 富集
kegg <- enrichKEGG(
gene = gene$ENTREZID,
keyType = "kegg",
organism = 'hsa',
pvalueCutoff = 1,
pAdjustMethod = "BH",
qvalueCutoff = 1,
use_internal_data = FALSE)
kegg2 <- setReadable(kegg, OrgDb = "org.Hs.eg.db", 'ENTREZID')
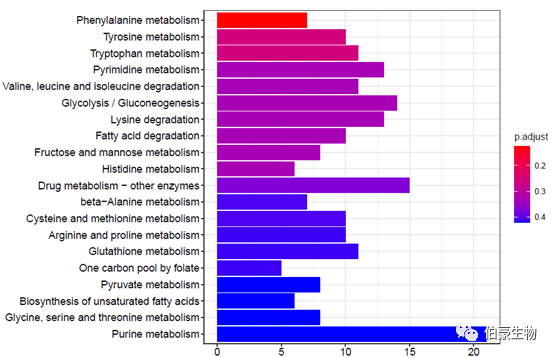
绘制柱状图:
barplot(kegg2,showCategory = 20)
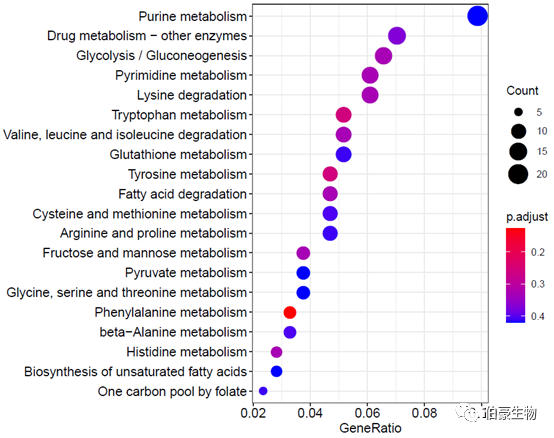
绘制气泡图:
dotplot(kegg2, showCategory = 20)
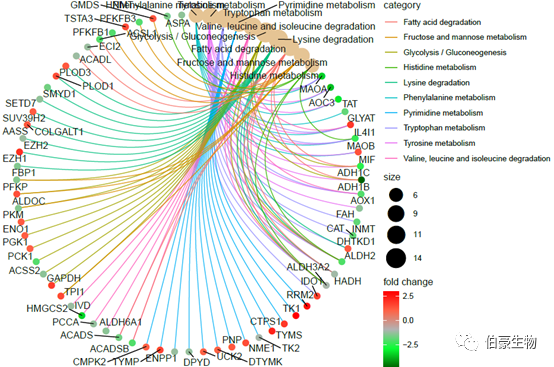
绘制网络图 1:
cnetplot(kegg2, showCategory = 10,foldChange=genelist, circular = TRUE, colorEdge = TRUE)
绘制网络图 2:
emapplot(kegg2, foldChange=genelist, showCategory = 20)

五、差异基因热图标记基因
当我们已经用自己的数据做过分析得到了差异基因,或者是已经有自己关注的基因,想用 TCGA & GEO 数据来验证自己的数据或结论的时候,用 TCGA & GEO 数据差异基因热图同时标记特定基因来展示结果就特别合适。
###### 差异基因热图标记关注基因
library("ComplexHeatmap")
library("circlize")
diff_exp = geneexp[rownames(diffgenes),]
# 进行 zscore 归一化
diff_exp_scaled = t(apply(diff_exp, 1, scale))
colnames(diff_exp_scaled) = colnames(diff_exp)
# 需要标记的基因
genes = c('CDC20','THRB','FAM13A','CCNA2','PRR15','CHI3L1','CLCA2','ABCA10','A2ML1','LCN2')
## 设置位置
gene_pos = which(rownames(diff_exp) %in% genes)
ha = rowAnnotation(foo = anno_mark(at = gene_pos, labels = genes))
m=round(max(abs(diff_exp_scaled)))
## 画热图
Heatmap(diff_exp_scaled, colorRamp2(c(-m,0,m),c("blue", "#EEEEEE", "red")),
right_annotation = ha,name = "Z-score",show_row_names =FALSE,show_column_names =FALSE)
绘图结果如下:

六、单个基因绘图
要在 TCGA & GEO 数据中验证自己的关注的基因的差异情况,除了前面说的差异基因热图标记特定基因之外,也可以对单个基因直接进行绘图。这里我们提供三种展示方式,选择自己喜欢的一种展示形式就行。
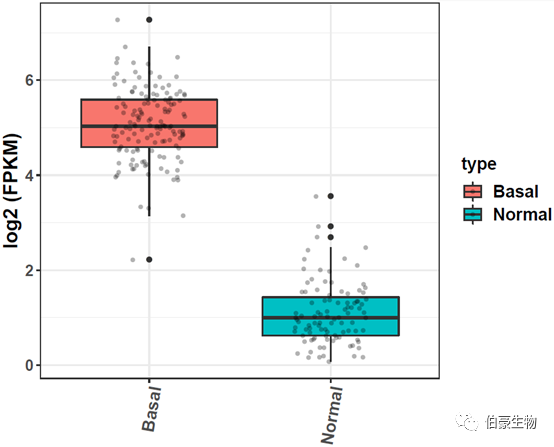
用箱线图展示:
## 单个基因箱线图
exp2 = data.frame(t(diff_exp))
exp2$type = group_file$type
ggplot(exp2, aes(x=type, y=CDC20,fill=type)) +
geom_boxplot()+geom_jitter(alpha = .3, width =0.2,size=1)+labs(title = "") + ylab("log2 (FPKM)")+ xlab("")+theme_bw()+
theme(title=element_text(face="bold",size=16),axis.title=element_text(face="bold",size=15),axis.text.x=element_text(face="bold",angle=80,size=13,hjust=1),
axis.text.y=element_text(face="bold",size=12),legend.text=element_text(face="bold",size=13),legend.title=element_text(face="bold",size=13))
结果如下:
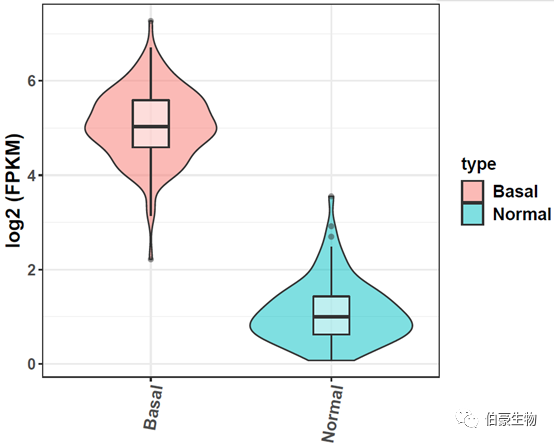
小提琴图展示:
### 单个基因小提琴图
ggplot(exp2, aes(x=type, y=CDC20,fill=type)) +
geom_violin(alpha=0.5) + geom_boxplot(alpha = .5,fill="white", width= .2)+labs(title = "") + ylab("log2 (FPKM)")+ xlab("")+theme_bw()+
theme(title=element_text(face="bold",size=16),axis.title=element_text(face="bold",size=15),axis.text.x=element_text(face="bold",angle=80,size=13,hjust=1),
axis.text.y=element_text(face="bold",size=12),legend.text=element_text(face="bold",size=13),legend.title=element_text(face="bold",size=13))
结果如下:
散点图展示:
### 单个基因散点图
ggplot(exp2, aes(x=type, y=CDC20)) +
geom_dotplot(binaxis='y', stackdir='center',binwidth = 0.1)+stat_summary(fun.y=median, geom="point", shape=18,size=3, color="red")+
theme(axis.title=element_text(face="bold",size=15),axis.text.x=element_text(face="bold",angle=80,size=13,hjust=1),axis.text.y=element_text(face="bold",size=12))
结果如下:
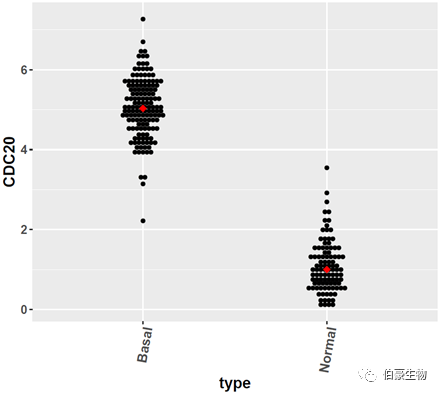
这里只展示一个基因的情况,如果是有多个基因需要绘图的,把代码里的基因名替换一下就好了。
好啦,今天的分享就到次为止啦,下次再继续哦!
更多伯豪生物人工服务:

