在发育过程中,细胞会对刺激做出反应,在整个生命过程中,从一种功能性“状态”转变为另一种功能性“状态”。拟时序分析(pseudo-time),它指通过构建细胞间的变化轨迹来重塑细胞随着时间的变化过程。用于拟时序分析的软件和算法很多,目前文章中用到 多的是 Monocle 软件,这也是是 CNS 主流期刊的常用软件。
对于单细胞的数据做拟时序分析基本的流程是挑选一些感兴趣并且可能有分化关系的亚群,然后来分析它们之间的分化关系。 那么对于空间转录组数据有没有什么新花样呢? 其实目前的空间转录组测序已发表的文章还没太注重细胞分化这一内容,可能大家做空转数据挖掘的时候还没太把关注点放到分化关系这上面吧,那么今天就来给大家写写自己的突发奇想吧!
既然是做的空间转录组,那么我们就要有效的利用起来空间位置信息。选择的亚群不能太离散。其次,我们可以分析相同细胞类型的亚群在空间位置的分化关系,也可以按照病理状态分布的渐进变化来选择区域做拟时序分化。
读取 seurat 对象
library(monocle)
# 读取前面保存的 seurat 对象文件
combin.data <- readRDS("combin.data.RDS")
这里我们选择大脑皮层区的亚群作为示例进行细胞分化分析。
# 展示亚群分布
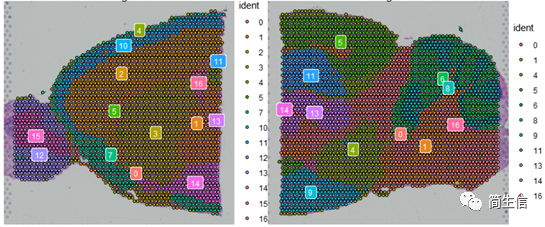
SpatialDimPlot(combin.data, label = TRUE, label.size = 3)
# 展示皮层 marker 的分布
SpatialFeaturePlot(combin.data, features = c("Stx1a"))
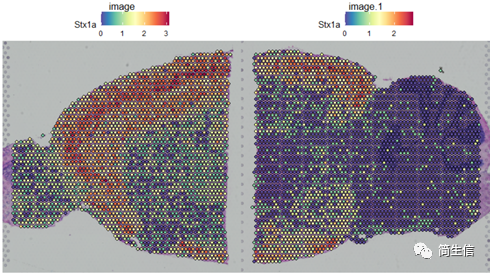
根据亚群和皮层 marker STX1A 的表达分布,我们选择 2、5、7、9、10 号群作为皮层的亚群。
# 选择要分析的亚群 subdata <- subset(combin.data, idents = c(2,5,7,9,10))
导入 seurat 对象,构建 CellDataSet 对象
这里我们使用 monocle2 做拟时序分析。monocle2 做拟时序分析需要三个矩阵文件:expression_matrix(表达矩阵,一般是 raw count)、phenoData(细胞 meta 文件,可以包括细胞的样本、亚群等信息)、featureData(gene 的 meta 文件,注意要包括 gene_short_name 这一列)。
# 单细胞 count 文件、细胞类型注释文件、基因注释文件
expression_matrix = combin.data@assays$Spatial@counts
cell_metadata <- data.frame(group = combin.data[['orig.ident']],clusters = Idents(combin.data))
gene_annotation <- data.frame(gene_short_name = rownames(expression_matrix), stringsAsFactors = F)
rownames(gene_annotation) <- rownames(expression_matrix)
##### 新建 CellDataSet object
pd <- new("AnnotatedDataFrame", data = cell_metadata)
fd <- new("AnnotatedDataFrame", data = gene_annotation)
HSMM <- newCellDataSet(expression_matrix,
phenoData = pd,
featureData = fd,
expressionFamily=negbinomial.size())
monocle2 做拟时序分析主要包括三个步骤:
1、基因筛选。选择要作为拟时序分化依据的基因,软件官方提供了几种可选的方法。
A、选择不同时间点分化差异基因,这应该是比较理想的情况,需要你提供的数据是根据不同分化时间点取样的;
B、选择表达离散度高的基因,也就是在所有细胞里变化比较大的基因,如果只是想看某个亚群里细胞之间的分化选择这种方法是比较合适的;
C、选择亚群间差异大的基因,一般想比较多个亚群间的分化关系选择这种方法效果会好一点;
D、选择一些已知跟分化相关的基因。
2、数据降维。Monocle2 使用 DDRTree 的非线性重建算法对细胞进行排序。
3、细胞排序。根据细胞的降维后的结果给每个细胞计算一个分化时间。不过这里有一点需要注意,做细胞排序这一步可以自己指定分化起点,要不然算法会随机选择一端作为起点,也就是说计算出来的分化时间有可能是倒过来的,即起点是终点,终点是起点。
# 评估 SizeFactors
HSMM_myo <- estimateSizeFactors(HSMM)
# 计算离散度
HSMM_myo <- estimateDispersions(HSMM_myo)
# 计算差异
diff_test_res1 <- differentialGeneTest(HSMM_myo,fullModelFormulaStr = '~clusters', cores = 4)
# 选择差异基因
ordering_genes <- subset(diff_test_res1, qval < 0.05)[,'gene_short_name']
# 基因过滤
HSMM_myo <- setOrderingFilter(HSMM_myo, ordering_genes)
# 降维
HSMM_myo <- reduceDimension(HSMM_myo, max_components=2, method = 'DDRTree')
# 细胞排序
HSMM_myo <- orderCells(HSMM_myo)
Monocle 结果可视化:
# 查看亚群分化关系
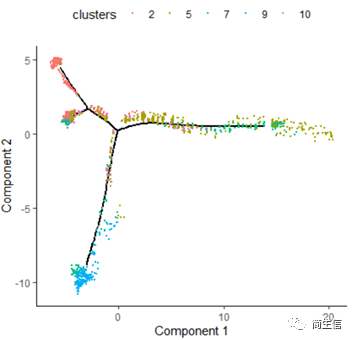
plot_cell_trajectory(HSMM_myo, color_by = "clusters",show_branch_points = F,cell_size =0.5)
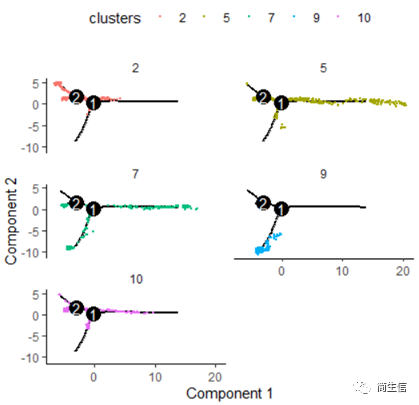
# 按亚群分开展示 plot_cell_trajectory(HSMM_myo, color_by = "clusters",cell_size =0.5) + facet_wrap(~clusters, nrow = 4)
# 分样本展示 plot_cell_trajectory(HSMM_myo, color_by = "orig.ident",show_branch_points = F,cell_size =0.5)
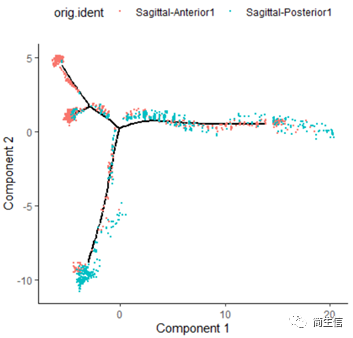
从亚群和样本的分布来看其实还是体现了一定的样本的异质性的,同一样本的亚群更容易分布到同一个分支上,这里的 2、7、10 号群有点分不太开,这时候我们就要思考一下,对于空转的数据有时候是不是单独分析某个样本上的分化是不是更合适一些。
选择单个样本的数据进行分析
这里我们选择小鼠大脑 Sagittal-Anterior1 的样本的皮层的亚群单独进行分析。
#### 单独绘制某个样本的图片
library("cowplot")
p1 <- SpatialPlot(combin.data,crop=FALSE,images='image')# 这里 Sagittal-Anterior1 样本的镜像图片的名字是 image,具体名称应跟前期读取镜像文件时设置的名字对应。
p2 <- SpatialFeaturePlot(combin.data, features = c("Stx1a"),images='image')
plot_grid(p1, p2)
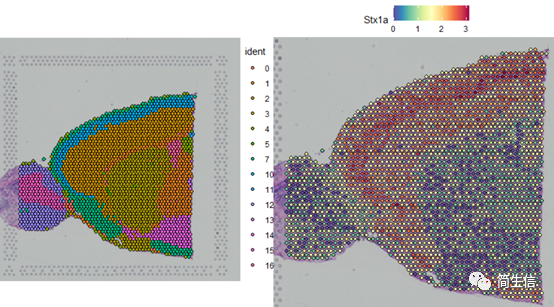
选择皮层 marker 高表达的 2、7、10 群进行拟时序分析。
# 选择单个样本 Sagittal-Anterior1 的亚群进行分析
subdata <- subset(combin.data, orig.ident == 'Sagittal-Anterior1')
subdata <- subset(subdata, idents = c(2,4,10))
# 单细胞 count 文件、细胞类型注释文件、基因注释文件
expression_matrix = subdata@assays$Spatial@counts
cell_metadata <- data.frame(group = subdata[['orig.ident']],clusters = Idents(subdata))
gene_annotation <- data.frame(gene_short_name = rownames(expression_matrix), stringsAsFactors = F)
rownames(gene_annotation) <- rownames(expression_matrix)
##### 新建 CellDataSet object
pd <- new("AnnotatedDataFrame", data = cell_metadata)
fd <- new("AnnotatedDataFrame", data = gene_annotation)
HSMM <- newCellDataSet(expression_matrix,
phenoData = pd,
featureData = fd,
expressionFamily=negbinomial.size())
# 评估 SizeFactors
HSMM_myo <- estimateSizeFactors(HSMM)
# 计算离散度
HSMM_myo <- estimateDispersions(HSMM_myo)
# 计算差异
diff_test_res1 <- differentialGeneTest(HSMM_myo,fullModelFormulaStr = '~clusters', cores = 4)
# 选择差异基因
ordering_genes <- subset(diff_test_res1, qval < 0.05)[,'gene_short_name']
# 基因过滤
HSMM_myo <- setOrderingFilter(HSMM_myo, ordering_genes)
# 降维
HSMM_myo <- reduceDimension(HSMM_myo, max_components=2, method = 'DDRTree')
# 细胞排序
HSMM_myo <- orderCells(HSMM_myo)
结果可视化:
# 查看亚群分化关系
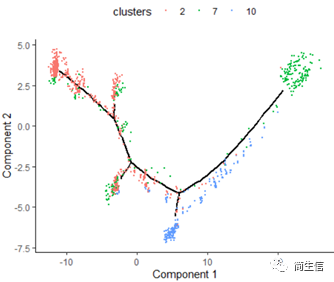
plot_cell_trajectory(HSMM_myo, color_by = "clusters",show_branch_points = F,cell_size =0.5)
# 按亚群分开展示
plot_cell_trajectory(HSMM_myo, color_by = "clusters",cell_size =0.5) + facet_wrap(~clusters, nrow = 4)
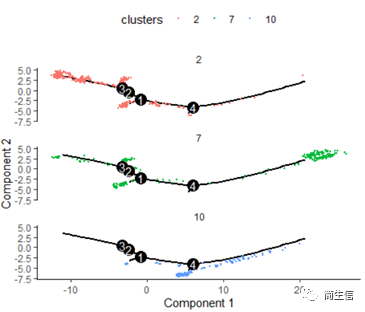
这里我们可以发现,之前选择两个样本一起的 5 个亚群分析的时候这 3 个群是有点区分不开的,在这次重新分析的时候却区分的很明显。这也说明选择的亚群范围不一样 monocle 得到的结果差异是很大的,究其原因可能是加入某些差异比较大的亚群或细胞后会进一步压缩原本差异比较小的亚群之间的差异分布。因为是算法同时计算多组差异变化难免会出现这种情况,所以我们在一开始选择亚群上就要稍微注意一点。
# 绘制分化时间
cell_Pseudotime <- data.frame(pData(HSMM_myo)$Pseudotime)
rownames(cell_Pseudotime) <- rownames(cell_metadata)
plot_cell_trajectory(HSMM_myo, color_by = "Pseudotime",cell_size =0.5)
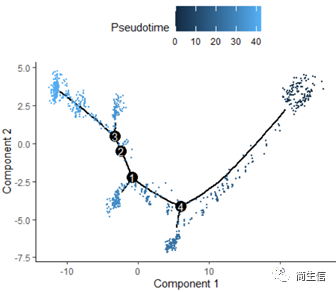
从分化时间的分布来看三个亚群的分化大概顺序是从 7 号群一 >10 号群一 >2 号群,由于 monocle 的判断的分化起点是随机的,所以也有可能是倒过来的顺序。
我们再来看一下皮层 marker 基因 Stx1a 的在分化过程中的表达分布
# 绘制 Stx1a 基因分化时间的变化
data_subset <- HSMM_myo['Stx1a',]
plot_genes_in_pseudotime(data_subset, color_by = "clusters")
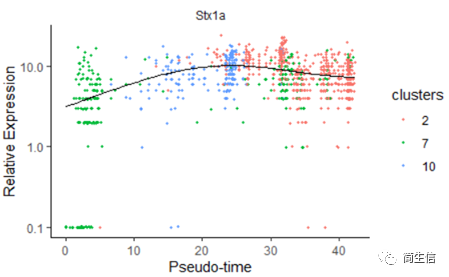
marker 基因的表达分布基本是由低到高再降低的趋势。
下面重点来了!我们把细胞的分化时间对应到组织切片上。
# 把分化时间对应到到组织切片位置上
combin.data[['Pseudotime']] <- 0
combin.data[['Pseudotime']][rownames(cell_Pseudotime),] <- cell_Pseudotime
SpatialFeaturePlot(combin.data, features = c("Pseudotime"),images='image')
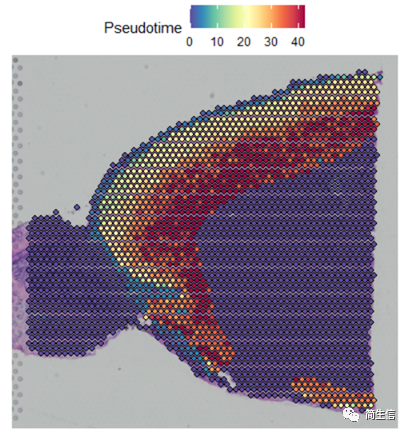
这时候我们就发现结果有点意思了,皮层细胞的分化在组织切片上是从外向内的方向的,当然也有可能实际上是反过来的,也就是从里往外的,总之就是细胞的分化是存在一定的空间位置规律的。
接着,我们再来看一下细胞分化过程中表达变化显著的基因有哪些。
# 分析分化时间显著变化的基因
diff_test_res2 <- differentialGeneTest(HSMM_myo[ordering_genes,],fullModelFormulaStr = "~sm.ns(Pseudotime)",cores = 4)
# 选择 top50 基因绘图
top50_gene = diff_test_res2[order(diff_test_res2$qval),][1:50,'gene_short_name']
plot_pseudotime_heatmap(HSMM_myo[top50_gene,], num_clusters = 6,cores = 1,show_rownames = T)
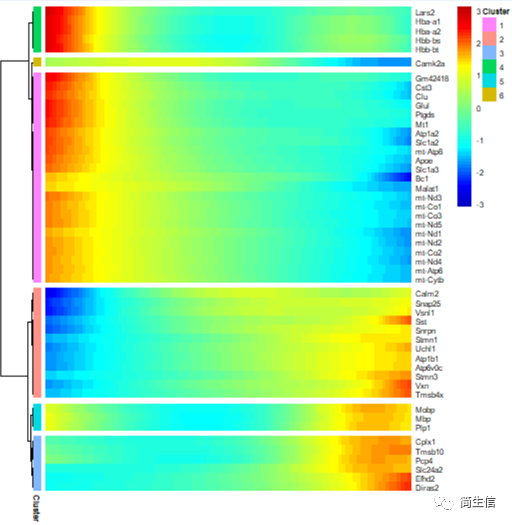

从 top50 差异基因的情况来看,血红蛋白基因和线粒体基因在小鼠大脑皮层切片外层的表达比较高。我们也可以把所有差异基因都输出来,将不同表达趋势的基因模块分别进行功能富集,这样就可以知道随着皮层细胞的在空间位置的分化哪些功能发生了变化。
sig_gene_names <- subset(diff_test_res2, qval < 0.05)[,'gene_short_name']
heatmap = plot_pseudotime_heatmap(HSMM_myo[sig_gene_names,], num_clusters = 6,cores = 1,show_rownames = F,return_heatmap=T)
###get cluster info
row_cluster = cutree(heatmap$tree_row,k=6)
write.table(row_cluster,file=paste("_gene_clusters.xls",sep=""),sep="\t",row.names=T)
细胞更有意思呢!因为示例数据里没有病理信息,如果结合病理特征来做拟时序分化分析应该能发现更多有价值的东西!
转载:简生信(侵删)

更多伯豪生物人工服务:

