染色体重排导致的基因融合事件广泛存在于多种肿瘤当中。例如在人类基因组研究中发现,异常的融合基因可引起恶性血液疾病以及肿瘤,包括 EMLA-ALK 和 BCR-ABL1 融合基因导致白血病,TMPRSS2-ERG(前列腺癌),EML4-ALK( 肺癌),VTI1A-TCF7L2( 直肠癌)[1]。由于基因融合产物是肿瘤中常见驱动因素,因此可作为抗癌治疗中潜在的预后和治疗靶标。小编之前着重介绍和比较了当下检测融合基因的相关技术,由于新一代测序(NGS) 技术的不断发展和广泛应用,利用生物信息学方法来检测融合突变也取得了较大的进展,因此本专题主要介绍 NGS 技术包括 RNA 和 DNA 两个层面及其相关的生信方法。
新一代测序检测融合基因主要有全基因组测序(WGS)、转录组测序(RNA-seq) 和目标区域靶向(根据靶向富集方法不同又分为杂交捕获和扩增子测序)等技术。一般而言,WGS 应该在合理的测序深度下进行融合基因检测,一般要求~30-60X,这对于肿瘤标本的低克隆性融合尤为重要。与之相反,RNA-seq 由于只能检测转录和剪接成熟 mRNA 的基因组区域,因此只能检测到相对高表达的融合。此外,根据已知的融合位点,设计特定的检测探针,采用靶向捕获 DNA 或 RNA panel 测序获得远高于 WGS 的测序深度,从而可以更敏感的检测出融合基因,因此该技术在临检领域更为适用。测序读长取决于 NGS 的平台,在检测融合基因时推荐较长的读长,从而尽可能捕获到断裂点 [2]。
检测技术 | 检测层面 | 主要实验过程 |
全基因组重测序(WGS) | DNA | 基因组 DNA 随机片段化,加接头,测序 |
目标区域杂交捕获测序 | DNA/RNA | 文库制备,加接头,特异性 DNA/RNA 序列富集,扩增后测序 |
转录组测序(RNA-seq) | RNA | mRNA 反转录 cDNA 并制备文库 |
目标区域扩增子测序 | RNA | 在融合点处扩增测序 |
核酸质谱 (MassArray) | RNA | 反转录,PCR,引物延展,基质辅助激光解吸 / 电离 |
数字化基因定量(NanoString) | RNA | 序列特异性杂交 |
融合基因检测常用基因组方法 [2]
RNA 测序技术能够很好地识别物种中产生异常变异的 RNA 种类,使发现因功能性或互作关系引起的基因融合及其导致的病变研究成为可能。双端配对的 RNA 测序能够提高基因的覆盖率,因此对检测基因融合具有特别的优势,该方法已被用于病理学的研究,并为调控与治疗提供潜在的可能性。目前已经有很多生物学工具基于高通量测序数据来检测融合基因,如 FusionSeq、deFuse、TopHat-Fusion、Fusion-Hunter、STAR-Fusion 等。
RNA 测序检测融合基因主要有两种分析策略:
1、基于序列比对的方法,寻找不一致序列(discordant pair reads) 和覆盖断裂点的序列(junction/split reads) 从而识别出融合事件。
2、基于拼接比对的方法,首先组装出新的转录本,然后比对到基因组,从而鉴定出与染色体重排一致的融合转录本 [3]。
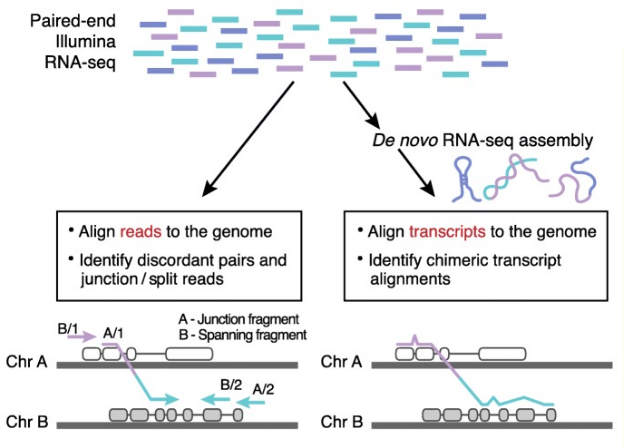
RNA-seq 检测融合基因的两种分析策略 [3]
目前针对 RNA-seq 已经开发出很多检测融合基因的生信工具,大多都是基于以上两种分析策略进行预测的。对于融合基因检测,RNA-seq 是一种非常高效的方法,可以在较低成本下对全基因组范围内的融合基因进行检测,其分辨率和通量比传统方法要高很多,并且还能发现新的融合基因。
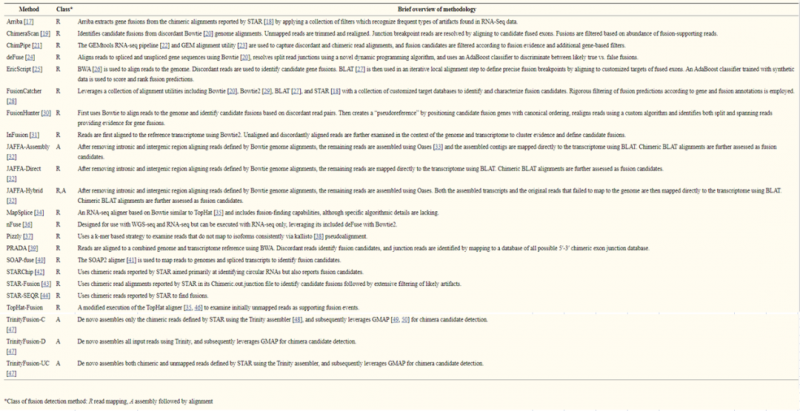
RNA-seq 检测融合基因工具汇总 [3]
分析工具测评结果显示,大多数情况下,STAR-Fusion 在所有检测工具中的排名高,其次是 Arriba 和 STAR-SEQR,见下图。在组装比对策略中,TrinityFusion- C 工具排名高。值得注意的是,排名前三的工具都是采用 STAR 工具进行比对的。
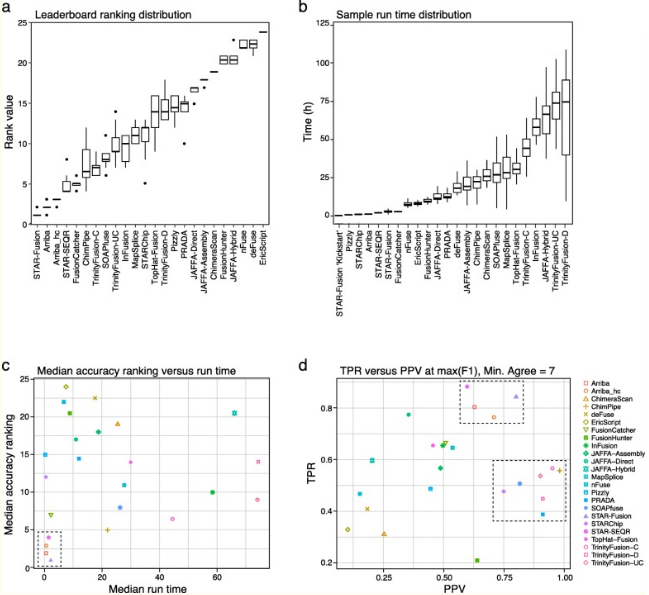
RNA-seq 分析融合基因工具准确性评估 [3]
DNA 测序也可以对融合基因进行检测,由于融合基因大多发生在内含子区域,采用 WGS 对整个基因组进行测序的方法往往成本较高,目前对于已知融合位点的检测,推荐目标区域靶向(panel) 测序。融合基因作为结构变异(SV) 的一种类型,采用 DNA 测序分析融合基因的策略主要包括:
1、双端序列配对法(pair-end method)
该方法总体思路是将双端序列比对到基因组上,然后评估双端配对序列的距离和方向是否和建库信息一致。
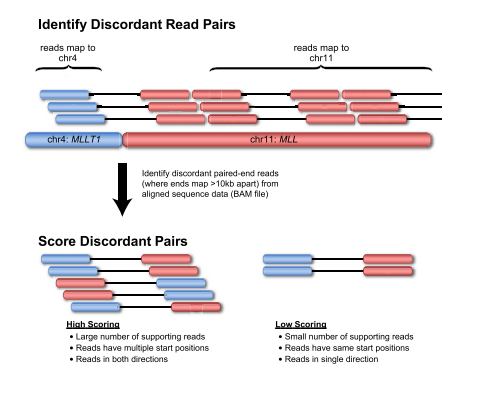
2、断裂序列法(split-read approach)
获得比对到基因组上的单端序列,将未能比对上的那部分序列切断,然后重新进行比对,获得比对位置和比对方向。
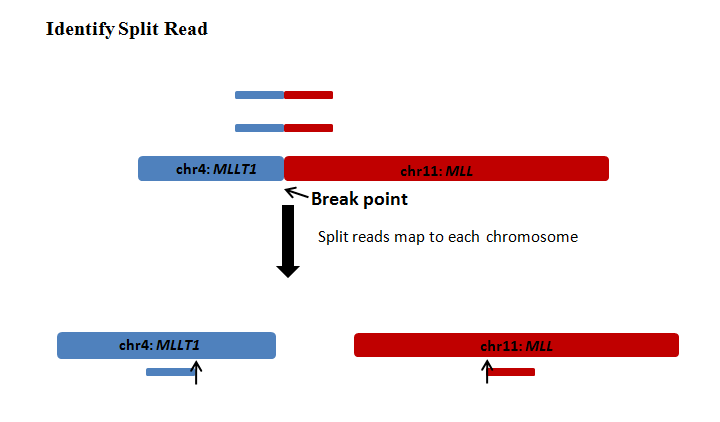
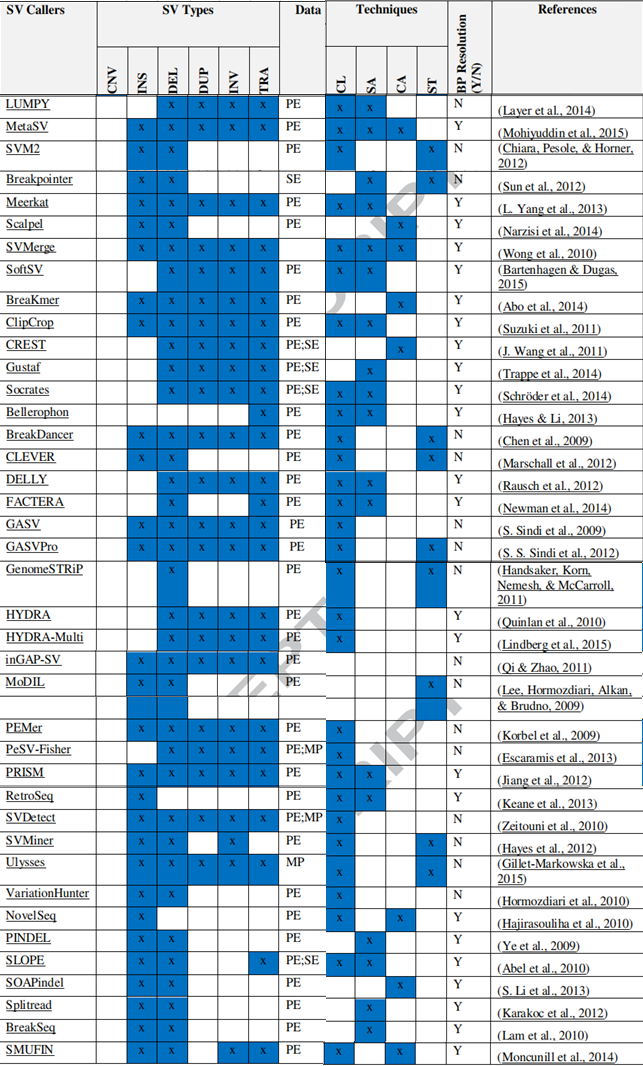
目前针对 DNA 测序开发出许多检测结构变异(SV) 的生信工具,见下图,也有专门检测融合基因的工具,比如 BreakID 和 GeneFuse 等工具。检测融合基因的工具大多基于以上两种分析策略进行预测的。不同的预测工具采用的策略有所不同,可以结合不同的检测工具来获得更好的灵敏度。当然,这种综合的方法并非是结果简单整合,而是优先考虑更准确的策略,例如断裂序列法要优先于双端序列配对法 [5]。
总的来说,过去的十几年里,新一代测序技术在融合基因检测方面发挥着越来越重要的作用,有着其他技术无法比拟的优势。随着测序技术的不断发展,产生的读长更长,也为融合基因的检测带来了新的机遇和挑战。
参考文献:
[1] Qingguo Wang, Junfeng Xia, Peilin Jia, et al. Application of next generation sequencing to human gene fusion detection: computational tools, features and perspectives. Brief Bioinform, 2013, 14(4): 506-519.
[2] Jan Schröder , Amit Kumar , Stephen Q Wong. Overview of Fusion Detection Strategies Using Next-Generation Sequencing. Methods Mol Biol, 2019:125-138.
[3] Brian J , Alexander Dobin, Bo Li, et al. Accuracy assessment of fusion transcript detection via read-mapping and de novo fusion transcript assembly-based methods. Genome Biol, 2019, 20(1): 213.
[4] Haley J and Eric Duncavage. Detection of structural DNA variation from next generation sequencing data: a review of informatic approaches. Cancer Genet. 2013, 206(12): 432-440.
[5] Peiyong Guan, Wing-Kin Sung. Structural Variation Detection Using Next-Generation Sequencing Data: A Comparative Technical Review. Methods, 2016,102:36-49.
更多伯豪生物人工服务:

